Frequently Asked Questions
- Is there a manuscript describing CancerGD?
- How should I interpret the effect size?
- How should I interpret the box plots?
- What dependencies are included?
- When is a driver gene considered 'altered'?
- Where does the genotyping data come from?
- Where can I get details of the genes considered altered in each cell line?
- Why are some dependencies absent from STRING?
- What does the Multiple Hit column indicate?
- How can I contribute a loss-of-function screen to CancerGD?
- How can I access this data from scripts, as JSON web service?
- How can I download the whole CancerGD database to interrogate locally?
Is there a manuscript describing CancerGD?
A manuscript describing CancerGD is available here.
How should I interpret the effect size?
The effect size presented in the database is the Common Language Effect Size, equivalent to the Probability of Superiority and the Area Under the ROC Curve. It indicates the probability that a cell line with an alteration in a particular driver gene will be more sensitive to a given RNAi reagent than a cell line without that alteration. For instance, the effect size in Campbell et al for the dependency of ERBB2 amplified cell lines upon MAP2K3 siRNA is 81%. This indicates that if two cell lines are chosen at random, one with ERBB2 amplification and one without, 81% of the time the cell line with the ERBB2 amplification will be more sensitive to the MAP2K3 siRNA.
Note that the Common Language Effect Size does not indicate the magnitude of the difference in sensitivity between the altered and wild-type group of cell lines. It merely indicates that one group is more sensitive than the other, not how much more sensitive.
What dependencies are included?
We include all dependencies that have a nominally significant p-value less than 0.05 and a Common Language Effect Size of greater than 65%. By nominally significant we mean that the p-value has not been corrected for multiple hypothesis testing. We, and others, have found that analysing all of the nominally significant dependencies associated with a driver gene can provide more insight than analysing only those that survive correction for multiple hypothesis testing.
How should I interpret the box plots?
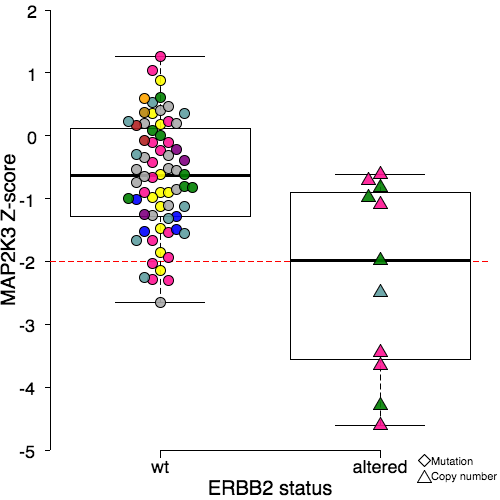
Each box plot shows the sensitivity of cell lines partitioned according to the status of a particular driver gene to RNAi reagents targeting a particular gene. For instance above we show the box plot indicating a dependency between ERBB2 amplification and sensitivity to MAP2K3 siRNA. The cell lines featuring the alteration in question are displayed on the right and the cell lines without the alteration are on the left. Each coloured object represents a cell line and the position along the y-axis indicates how sensitive that cell line is to the RNAi reagents targeting the gene indicated. The colours indicate the tissue of origin for each cell line, as indicated in the legend. The shape of each object indicates the alteration type (circle = no alteration, triangle = copy number, diamond = mutation). The boxes themselves are Tukey box plots – the bold line indicates the median of each group, the edges of the box indicates the interquartile range, and the whiskers indicate 1.5 times the interquartile range.
When is a driver gene considered 'altered'?
We integrate copy number profiles and exome sequencing to identify likely functional alterations in cancer driver genes. For most oncogenes we consider a functional alteration to be either an amplification, recurrent missense mutation, or recurrent in frame deletion or insertion. Recurrence is defined as as at least 3 previous mutations of a particular site in the COSMIC database. For a small number of oncogenes (ERBB2, MYC, MYCN) we consider only amplifications as functional events, while for another group (KRAS, BRAF, NRAS, HRAS) we only consider recurrent mutations/indels. In addition to recurrent missense or indel events, for tumour suppressors we consider that all nonsense, frameshift and splice-site mutations are likely functional alterations. We also consider that deletions (derived from copy number profiles) are functional alterations for tumour suppressors.
Where does the genotyping data come from?
Exome data for ~1,000 cell lines are obtained from the GDSC resource. Copy number data for the same set of cell lines is obtained from CancerRXGene. We use the gene level copy number scores which are derived from PICNIC analysis of Affymetrix SNP6.0 array data. An oncogene is considered amplified if the entire coding sequence has 8 or more copies while a tumour suppressor is considered deleted if any part of the coding sequence has a copy number of 0.
Where can I get details of the genes considered altered in each cell line?
The alteration matrix, indicating whether or not each driver gene is altered in each cell line, can be downloaded here.
Why are some dependencies not sent to STRING?
A maximum of 300 genes are sent to the STRING database for drawing the dependencies network. If more then 300 genes are present in the dependencies table, then the top 300 highest ranked genes will be sent to STRING. By default this means the 300 genes with the lowest associated p-values will be sent to STRING, but it is possible to sort the table based on other columns using the toggles in the column headers. This enables the use of STRING to visualize the 300 dependencies with the highest effect sizes or the biggest difference in scores.
What does the Multiple Hit column indicate?
The Multiple Hit column indicates whether a specific dependency has been observed in more than one dataset. For example, in breast cancer, ERBB2 amplification has been associated with an increased dependency upon ERBB3 in three individual screens (Campbell et al 2016, Marcotte et al 2016, Cowley et al 2014) so is indicated with a Yes in the Multiple Hit column. Note that a dependency is only considered a Mulitple Hit if the same driver was associated with the same dependency in the same cancer type (or Pan-Cancer in both screens). A dependency seen in one dataset in lung cancer and in another in breast cancer would not be considered a Multiple Hit. You can download all multiple hit dependencies from the 'Downloads' page
How can I contribute a loss-of-function screen to CancerGD?
We would be delighted to host additional loss-of-function screens in CancerGD. We have a preferred format for screens (outlined below) but will be happy to work with you to get your data into CancerGD. Click here to send us a mail.
To enable easy inclusion of future screens in CancerGD we request that data be provided as a tab-delimited table with each row representing a particular cell line and each column representing reagents targeting a specific gene. Cell line names should preferably follow the Cancer Cell Line Encyclopaedia naming convention (NAME_TISSUE all in uppercase) but COSMIC IDs are also acceptable. Genes should preferably be identified using ENTREZ IDs but other unique IDs (ENSEMBL Gene IDs) are acceptable. Due to regular changing and updating, gene symbols alone should not be used as unique gene identifiers. We favour SYMBOL_ENTREZID as gene IDs (e.g KRAS_3846) for ease of use but this is not required. In cases where multiple distinct scores are provided for a specific gene, as happens with scores from the ATARIS algorithm, we request that they be identified using distinct suffixes (e.g. KRAS_3846_1, KRAS_3846_2). Individual entries in the table should be quantitative scores indicating how sensitive a specific cell line is to perturbation of a particular gene. As different scoring procedures are used to quantify the results of screens using different experimental approaches (e.g. ATARIS (Shao et al., 2013) and zGARP (Marcotte et al., 2012) for shRNA screens, Z-score for siRNA screens (Campbell et al., 2016)) we do not require the scores to be in any standard format or range. However, we follow the convention in the field and suggest that increasingly negative scores should indicate greater inhibition of cell growth. A sample screen from Campbell et al (Campbell et al., 2016) is provided in the appropriate format here.
How can I access this data from scripts as a JSON web service?
We are still developing the JSON API and welcome feedback or requests for alternative features. Click here to send us a mail.
(1) This dependency data can also be obtained in JSON format for input into scripts. The data in the dependency table can be obtained, using a URL of the format:
http://www.cancergd.org/get_dependencies/driver/12345/TISSUE/STUDY/where:
- "driver" to indicate search is by driver gene. Change this to "target" to search by target gene.
- "12345" is the Entrez_ID of the gene gene (eg. 2064), as listed on the drivers page or targets page.
- "TISSUE" is either: "ALL_HISTOTYPES", or one of the tissues (eg. LUNG) listed on the tissues page.
- "STUDY" is either "ALL_STUDIES", or one of the study PubMed Id's as listed on the studies page.
For example:
(a) To retreive data for driver gene ERBB2, all tissues and all studies:
http://www.cancergd.org/get_dependencies/driver/2064/ALL_HISTOTYPES/ALL_STUDIES/
(b) To retreive data for target gene ERBB2, Pan-cancer, study Campbell(2016):
http://www.cancergd.org/get_dependencies/target/2064/PANCAN/26947069/
You can retrieve and extract the data from JSON, using a script. In Python 3, you could use:
import urllib.request, json
url = "http://www.cancergd.org/get_dependencies/driver/2064/BREAST/26947069/"
response = urllib.request.urlopen(url).read().decode('utf-8')
data = json.loads(response)
This data contains similar dependency information as the 'Download as CSV' link above the dependency table.
(2) The drivers can be obtained in JSON format using the URL.
http://www.cancergd.org/get_drivers/
Optionally a '?name=GENE_NAME' can be added to retreive only those drivers containing this string, eg:
http://www.cancergd.org/get_drivers/?name=ERwhich would retreive drivers containing 'ER', eg: ERBB2, RERE, DICER1, etc.
(3) You can also retrieve information about one gene, by using:
http://www.cancergd.org/get_gene_info/ERBB2/
(4) You can also retrieve the boxplot data in CSV (comma-separated-value) format using 'get_boxplot' followed by driver, target, tissue and study-pubmed-id:
http://www.cancergd.org/get_boxplot/csv/ERBB2/MAK2P/PANCAN/26947069/
How can I download the whole CancerGD database to interrogate locally?
The full CancerGD database can be downloaded from the 'Downloads' page as an Sqlite3 database or as a Comma-separate values (CSV) file.
To query the SQLite database you can use a program such as the free SQLite Browser